|
|
|
|
|
|
|
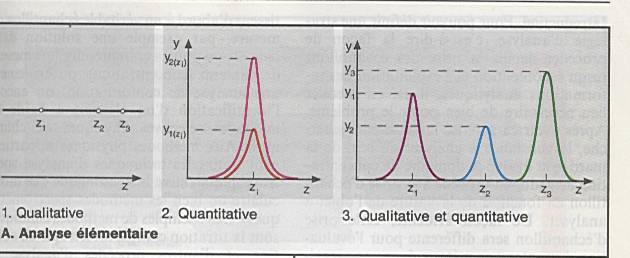
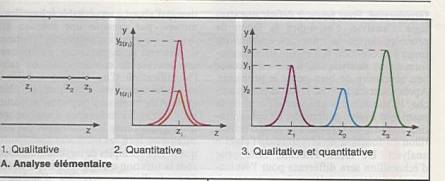
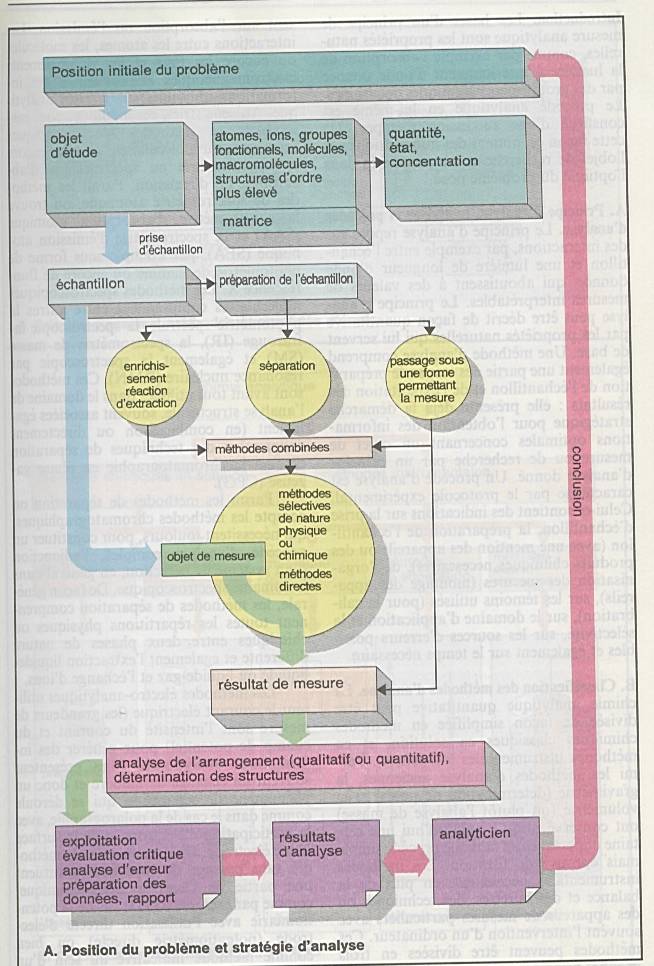
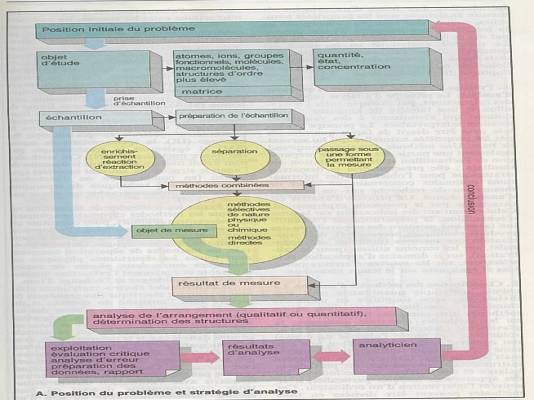
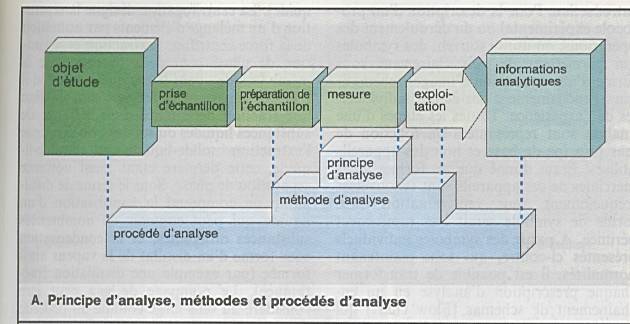
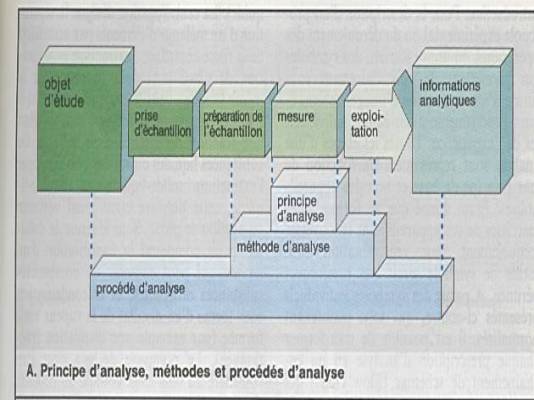
Poser le problème : que recherchez-vous ?
Une
substance à analyser au sein d ’une matrice. |
|
Evaluer le domaine de concentration. |
|
Prise d’échantillon |
|
|
|
|
|
|
|
|
Séparation de la fraction gênante de la matrice |
|
Séparation de plusieurs substances pour ensuite
faire l’analyse de l’une d’entre elles. |
|
Les techniques de séparations reposent sur des
techniques qui ne modifient pas les substances : précipitation,
extraction,.. |
|
On aboutit ainsi aux échantillons de mesures. |
|
|
|
|
Méthodes physiques : colorimétrie,
spectrophotométrie, fluorimétrie, .. |
|
Méthodes chimiques : complexométrie. |
|
|
|
Les différentes méthodes aboutissent à un
résultat de mesure |
|
|
|
|
Composition de l’échantillon |
|
Présence et concentration d’un ou plusieurs
éléments |
|
Validation des appareillages |
|
Examen critique des résultats : limite de
détection et de quantification |
|
Analyse des erreurs : validation des données |
|
Présentation des résultats : tableau, graphique,
paramètres statistiques |
|
|
|
|
|
|
|
|
Repose sur des interactions comme par exemple
entre l’échantillon et une lumière de longueur d’onde précise. |
|
Aboutissent à des mesures interprétables |
|
Ces principes sont décrits par les propriétés
naturelles. |
|
|
|
|
|
|
|
|
|
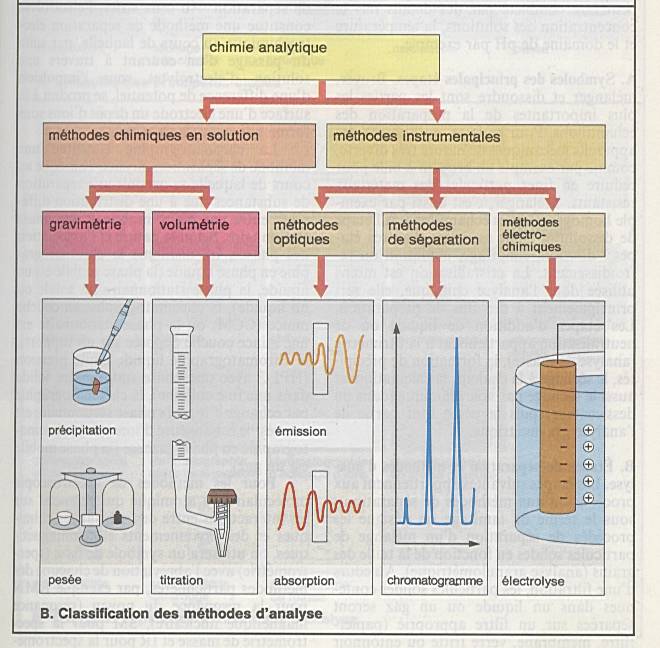
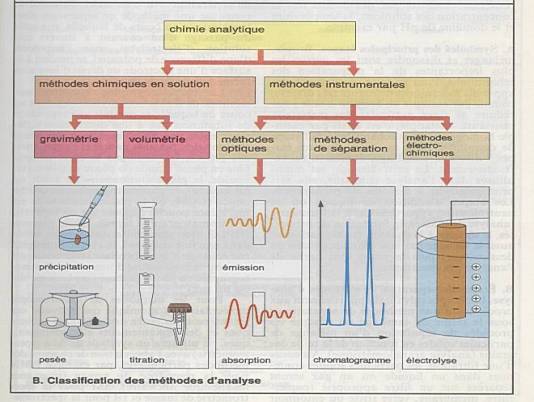
Méthodes chimiques classiques : gravimétrie,
volumétrie, titration. |
|
Méthodes instrumentales modernes : |
|
Méthodes optiques : principe d’analyse :
l’absorption ou l’émission. |
|
Méthodes chromotypographies : HPLC |
|
Méthodes électron-analytiques |
|
|
|
|
|
|
|
Le facteur limitant pour le choix de la méthode
d’analyse est la quantité d’échantillon et le domaine de concentration. |
|
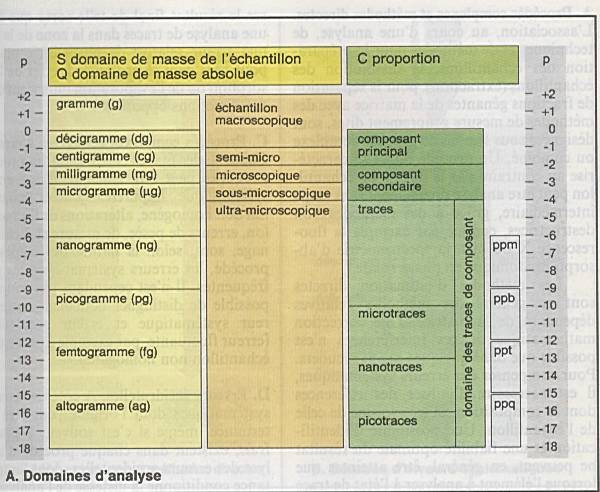
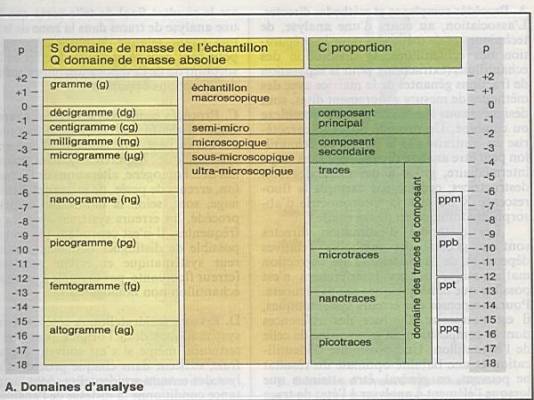
IUPAC en 1979 a défini 3 grandeurs fondamentales
: |
|
Le domaine de masse S |
|
Le domaine de masse absolue Q |
|
Le domaine de proportion P |
|
|
|
|
Le domaine de masse de l’échantillon : S=mx+my
ou x représente le composant ou l’analyte et y la matrice |
|
Les quantités les plus utilisées vont du gramme
(g) au microgramme (µg). |
|
Pour la quantité d’échantillon, on parlera
d ’échantillons macro, semi-micro, micro, sous-micro et
ultramicroscopiques |
|
En dehors du gramme, on utilise comme unité le
millilitre (ml) ou la mole (M). |
|
|
|
|
La notion de domaine de masse absolue Q,
correspond à la quantité de l'analyte x pour le dosage duquel on doit
utiliser un procédé d'analyse. |
|
|
|
|
|
|
La
proportion C est donnée par le quotient de la quantité de l'analyte x et de
la somme mx+ la masse de la matrice my, c'est-à-dire
la masse totale de l'échantillon. |
|
En fonction de la proportion on distinguera : |
|
Les analytes principaux de 100% à 10% |
|
Les analytes secondaires : de 10 à 0.1% |
|
Les traces : en dessous de 0.1% : |
|
ppm : partie par million |
|
ppb : partie par billion |
|
ppt : partie par trillion |
|
|
|
|
|
|
|
|
|
|
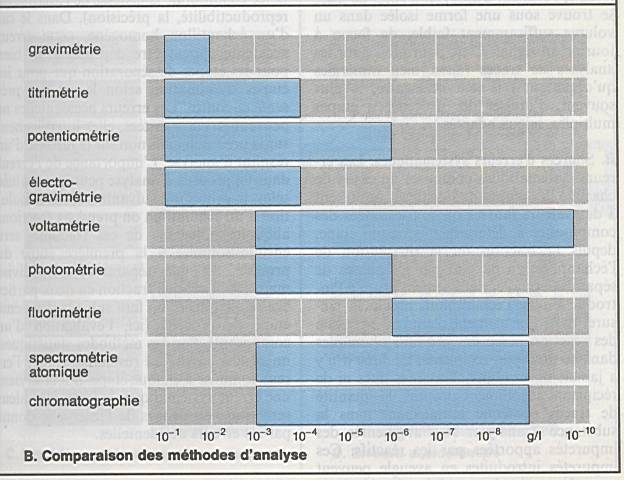
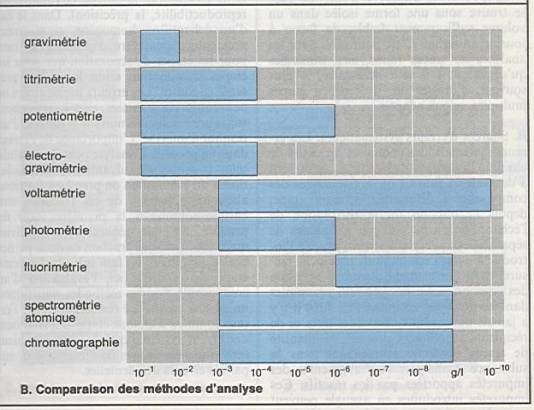
Titrimétrie : 10-2 à 10-4
g/l |
|
Potentiométrie : µg/l |
|
Photométrie : 10-3 à 10-6g/l |
|
Fluorimétrie : 10-6 à 10-9g/l |
|
Chromatographie : 10-3 à 10-9
g/l |
|
|
|
|
|
|
|
|
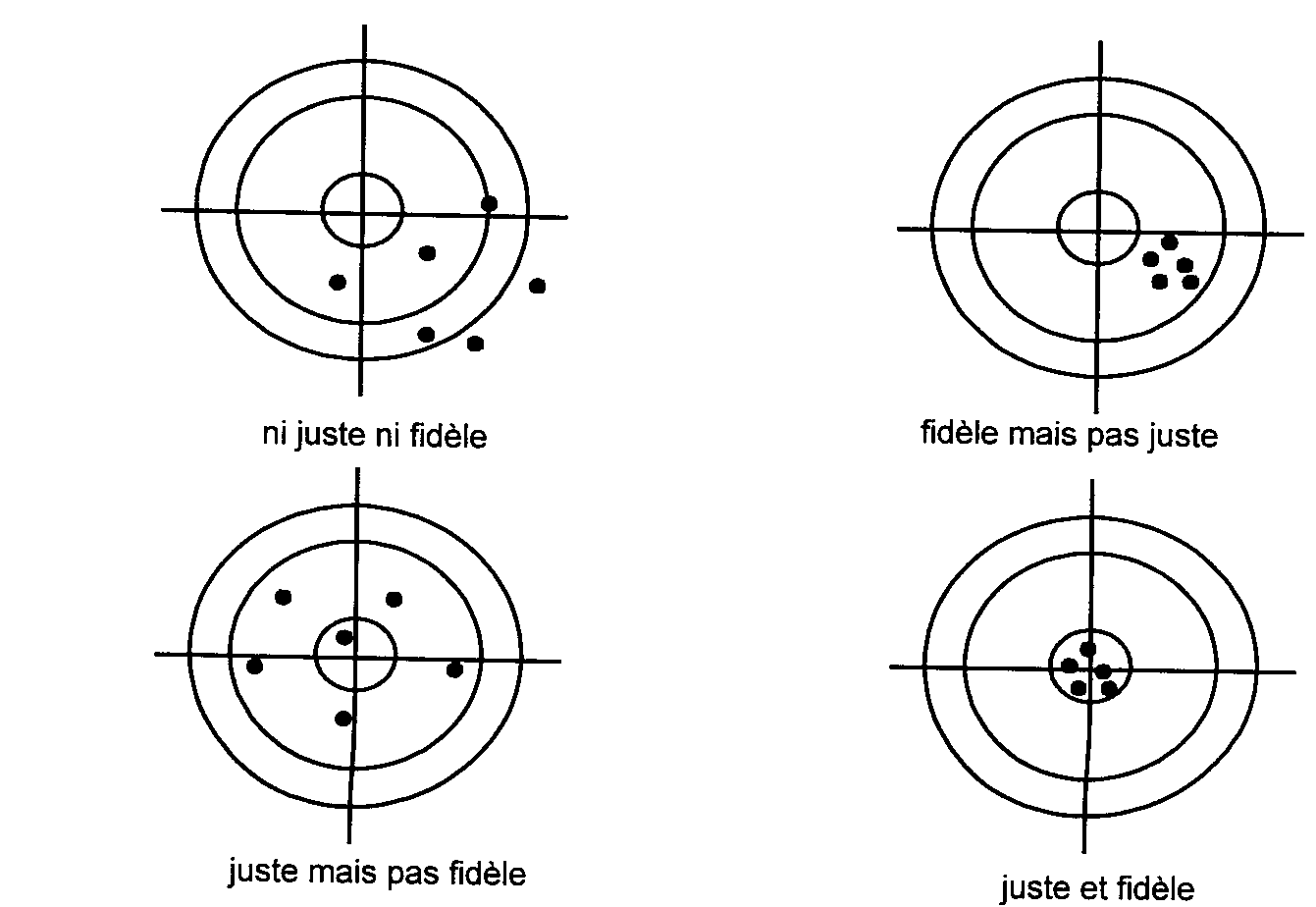
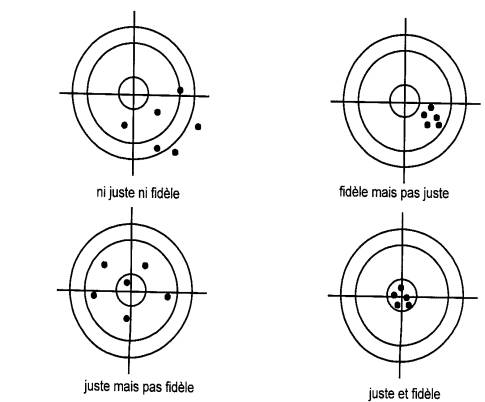
Fidélité : étroitesse de l ’accord entre
les mesures effectuées sur des prises multiples d ’un échantillon
homogène. |
|
Répétabilité : mesure de la fidélité lorsque les
mesures sont faites par un même opérateur, un même instrument, selon une
méthode unique et dans un délai court. |
|
Reproductibilité : mesure de la fidélité sans
restriction d ’opérateur, d ’instrument, de méthode ou de temps. |
|
|
|
|
Justesse : étroitesse de l ’accord entre
une mesure ou la moyenne des mesures et la valeur conventionnellement vraie
de l ’échantillon. |
|
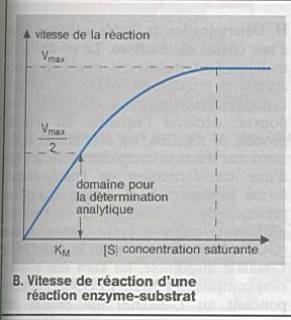
Sensibilité : rapport de la variation de la
réponse instrumentale à la variation de la concentration. |
|
Spécificité : mesure de l’analyte avec la
garantie que le signal ne provient que de l’analyte |
|
|
|
|
|
|
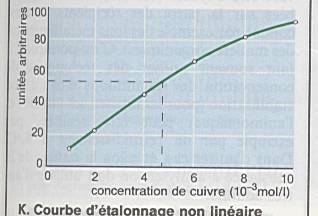
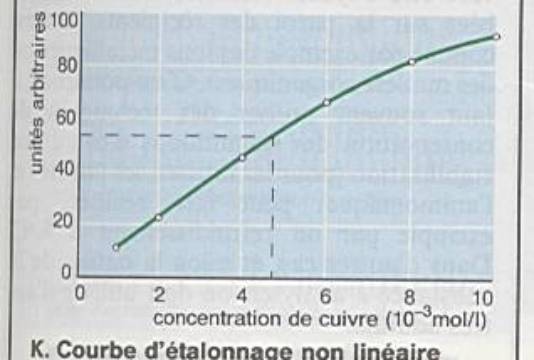
Linéarité : capacité à fournir des réponses
proportionnelles à la concentration en analytes à doser |
|
Limites de linéarité : limites expérimentales
entre lesquelles un modèle d ’étalonnage linéaire peut être appliqué. |
|
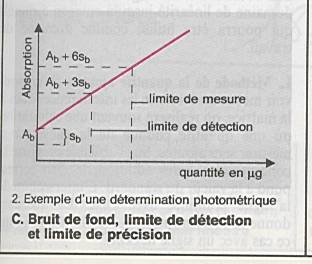
Limite de détection : la plus petite concentration de l ’analyte pouvant être
détectée avec un risque d ’erreur connu. |
|
|
|
|
|
|
Limite de quantification : la plus petite
concentration de l ’analyte pouvant être quantifiée avec un risque
d ’erreur connu. |
|
Capacité de mesure : rapport entre la fidélité
de la méthode et la dispersion des échantillons. |
|
Robustesse : importance des effets observés
lorsqu ’on applique de légères variations contrôlées. |
|
|
|
|
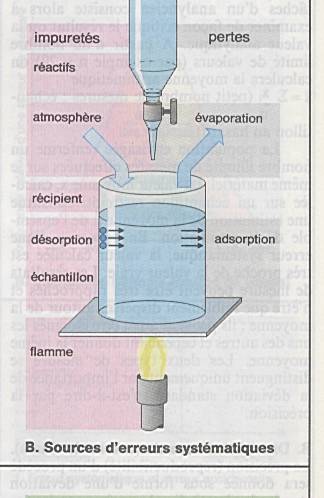
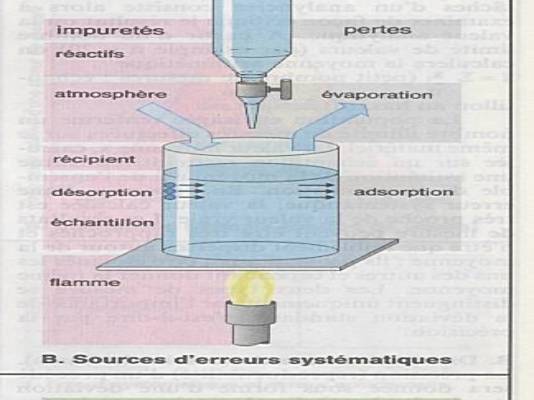
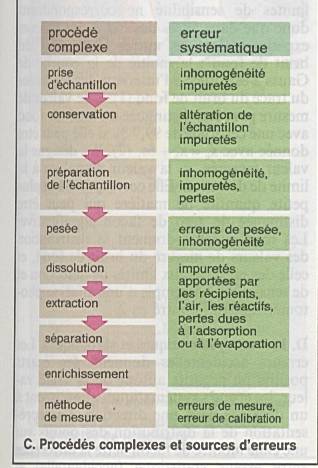
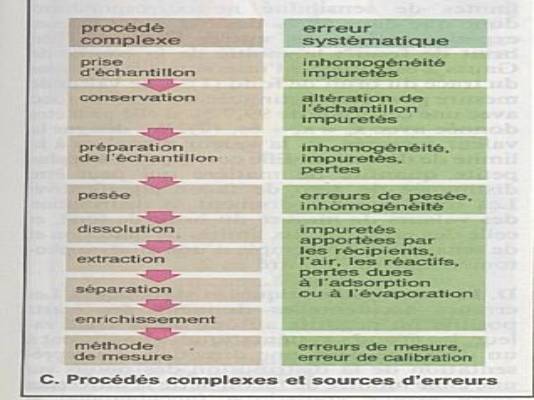
Ce sont les erreurs les plus fréquentes |
|
Il n’y a pas de produits chimiques absolument
purs ou de récipients absolument propres.. |
|
Impuretés ajoutées, échantillon non homogènes,
altération de l’échantillon, erreurs de pesée, de mesure, d’étalonnage,.. |
|
Ces impuretés introduites en aveugles peuvent
fausser le résultat final. |
|
Erreurs qui conduisent à une surestimation ou
une sous-estimation des composants. |
|
|
|
|
|
|
|
|
|
|
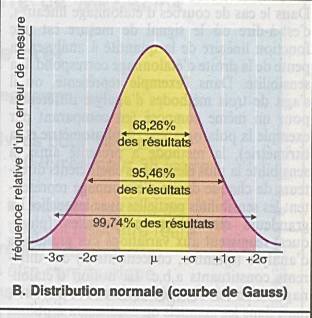
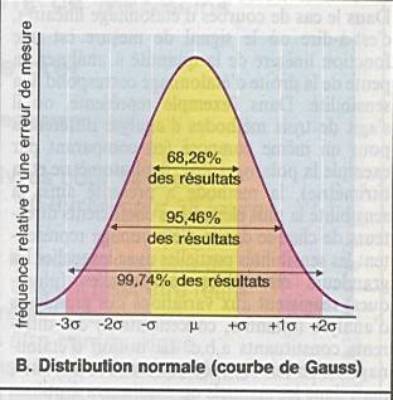
Elles interviennent sur la précision mais non
sur la justesse d’un résultat. |
|
On peut la réduire en répétant les expériences. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
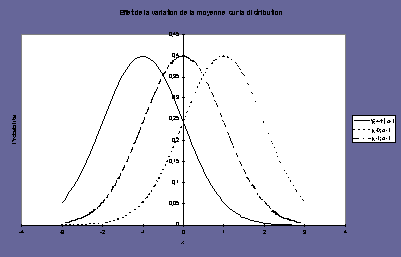
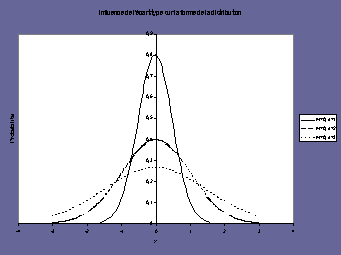
la moyenne (mean) : m |
|
l’écart-type
(standard deviation) : s |
|
le coefficient de variation ( coefficient of
variation) : C.V. en % |
|
|
|
|
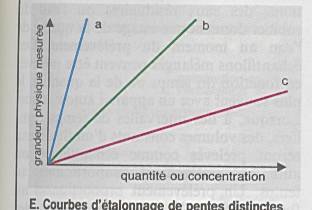
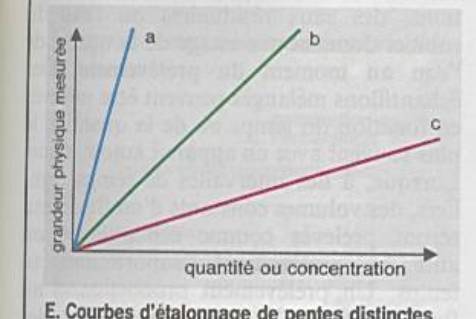
Le signal de la mesure est une fonction linéaire
de la quantité à analyser. |
|
La pente représente la sensibilité |
|
|
|
|
|
|
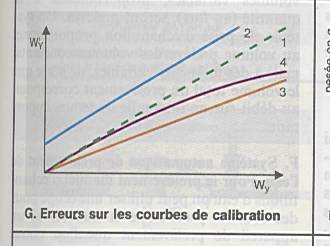
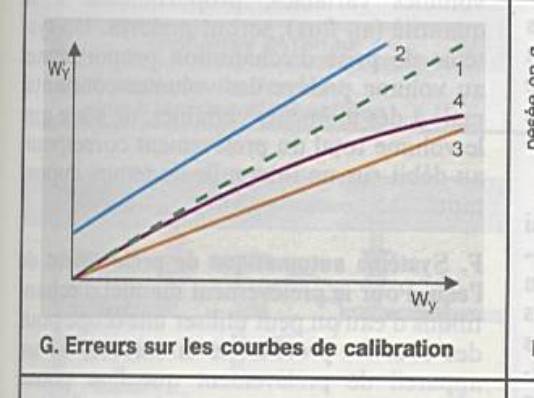
L’interférence due à la matrice |
|
Etalon inapproprié |
|
|
|
|
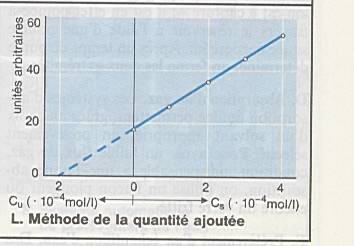
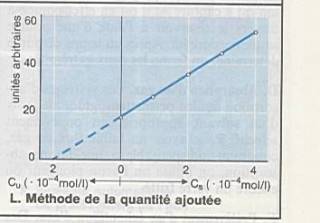
Peut être corrigé par la méthode de la quantité
ajoutée : |
|
|
|
|
|
|
|
|
|
|
|
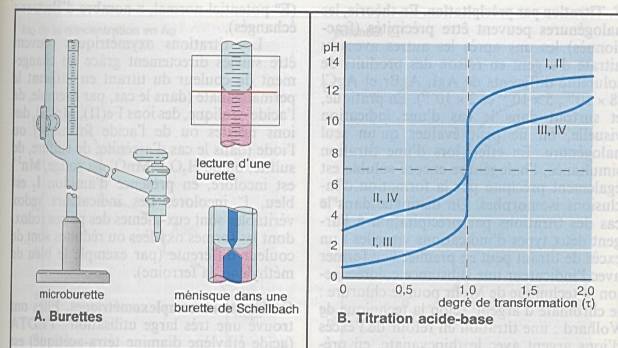
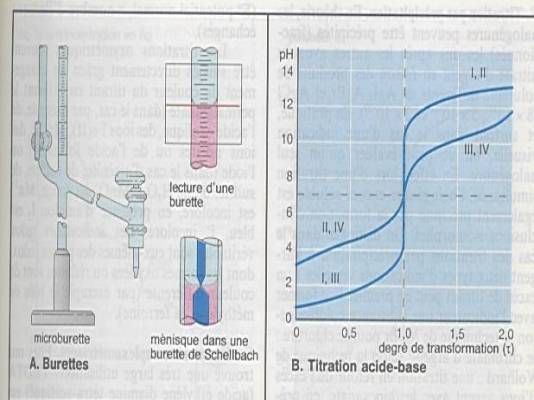
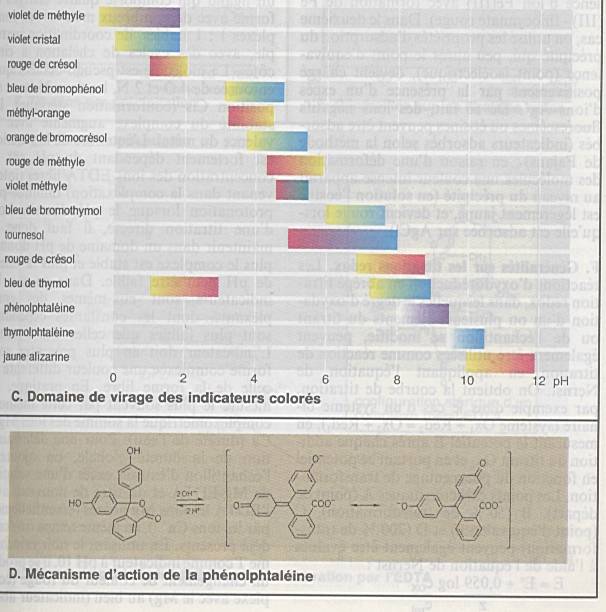
Interprétation des analyses titrimétriques
s’effectue à partir des courbes de titration |
|
Importance de l’allure de la courbe pour la
précision |
|
Acide fort-base forte : pH 4-10 |
|
Acide faible –base forte : pH 7-10 |
|
Acide fort – base faible : autour de 7 |
|
Acide faible – base faible : pH 4-7 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
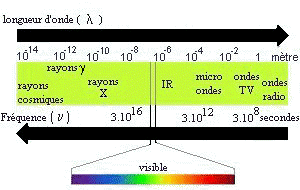
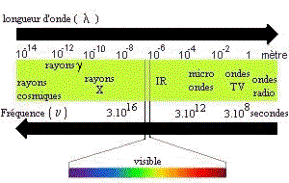
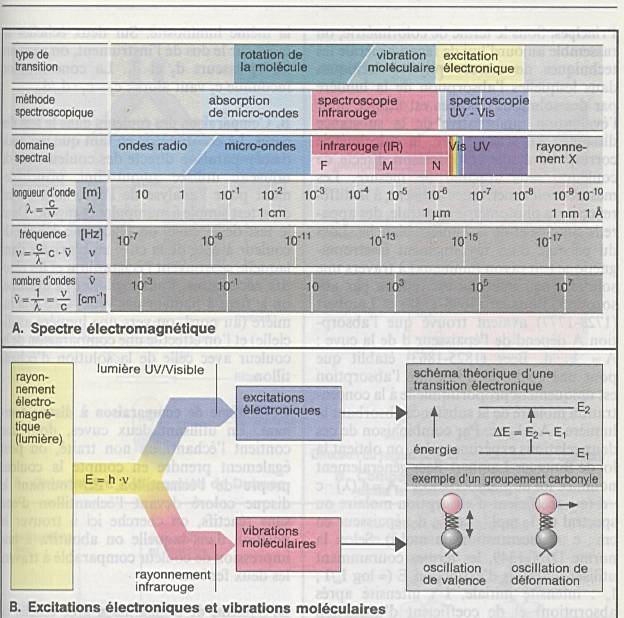
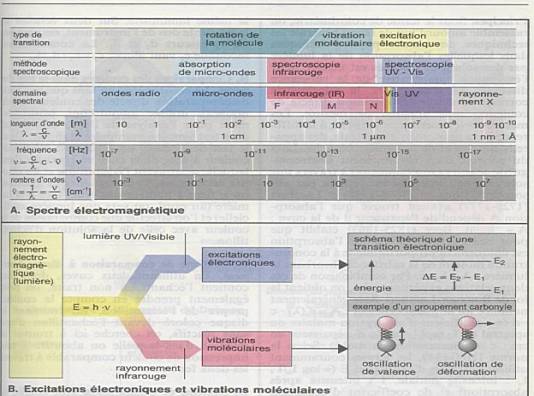
Ensemble continu des ondes électromagnétiques connues,
classées dans l'ordre de leurs longueurs d'ondes, de leurs fréquences, de
leurs énergies. |
|
|
|
|
longueur d'onde l : unité de longueur (distance)
qui sépare deux pics d'onde (exprimé en nm). |
|
nombre d'onde s : nombre d'onde par unité de
longueur, (exprimé généralement en cm-1), |
|
fréquence n : nombre d'onde par seconde (exprimé
en Hz). |
|
où c la célérité est la vitesse de la
lumière. |
|
|
|
|
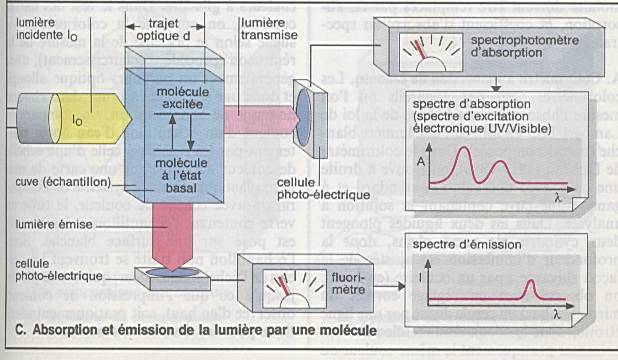
Io est l'intensité de la lumière
incidente |
|
Ir celle de la lumière réfléchie |
|
Ia celle de la lumière absorbée, |
|
It celle de la lumière transmise, |
|
Io = Ir + Ia +
It |
|
|
|
|
Loi de Bouguer – Lambert : l'intensité de la
lumière émergente I décroît exponentiellement lorsque l'épaisseur l du milieu
absorbant augmente : |
|
Loi de Beer:
où e est un coefficient
d'absorption caractéristique de la substance appelé coefficient d'extinction,
l est l'épaisseur de la cuve et c la concentration de la solution. |
|
|
|
|
|
|
Loi de Beer-Lambert : loi fondamentale : |
|
Si la concentration est exprimée en grammes par
litre, e est appelé coefficient d'extinction spécifique. |
|
|
|
Si la concentration est exprimée en moles par
litre, e est appelé coefficient d'extinction molaire. |
|
|
|
|
La densité optique D.O. ou D, encore appelée
extinction, est le logarithme du rapport de l'intensité incidente à
l'intensité transmise. |
|
|
|
La transmission T est définie comme le rapport
de l'intensité transmise à l'intensité incidente |
|
|
|
|
|
|
|
|
Soit l'ionisation, soit de la dissociation, soit
de l'association de solutés. |
|
Lorsque le soluté forme des complexes, la
composition de ces derniers dépendant de la concentration, |
|
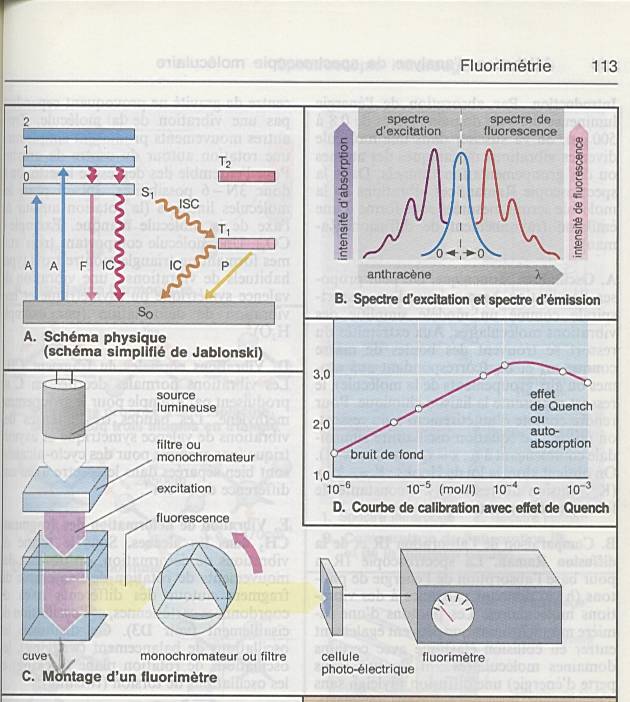
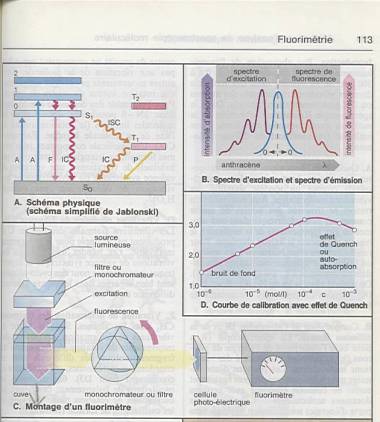
Lorsqu’il s'agit de solutions fluorescentes ou
de suspensions. |
|
Pour vérifier la loi, il faut une lumière
monochromatique |
|
|
|
|
|
|
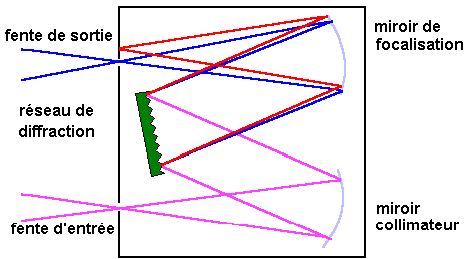
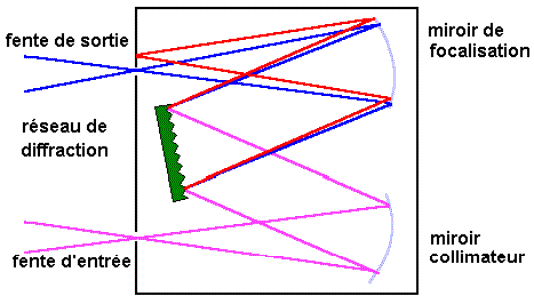
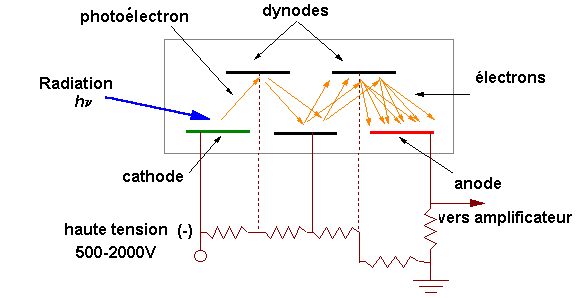
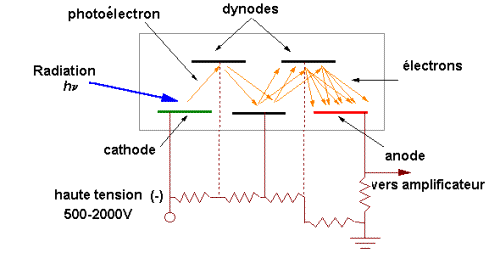
Une lampe à décharge au deutérium utilisée dans
le domaine de 190 à 400 nm |
|
|
|
|
|
Une lampe à filament de tungstène pour la région
allant de 350 à 800 nm. |
|
|
|
|
|
|
|
|
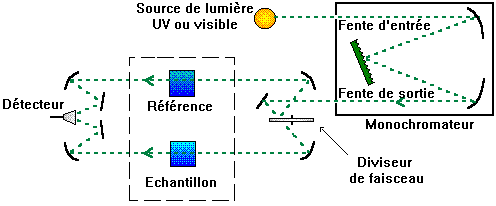
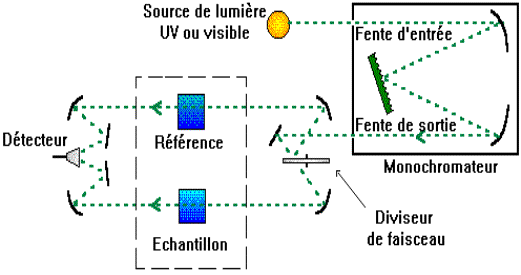
Elle contient soit l'échantillon soit la
référence. La longueur de la cuve est définie |
|
Elle doit être transparente aux radiations
d'étude. |
|
Par exemple en UV, les cuves sont en quartz,
elles ne peuvent être ni en verre ni en plastique. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
INJECTION : assurer l'introduction correcte de
l'échantillon dans le système |
|
SÉPARATION : séparer les composants d'un mélange
à l'intérieur d'une colonne |
|
DÉTECTION : détecter les composants lorsqu'ils sortent du
système. |
|
|
|
|
|
|
|
Selon la phase : |
|
Adsorption |
|
Partage |
|
Echange d'ions... |
|
Selon l’état de la phase mobile : |
|
Liquide |
|
Gaz |
|
Selon type d'appareillage utilisé |
|
chromatographie sur papier |
|
chromatographie sur couche mince |
|
chromatographie en phase liquide basse pression |
|
chromatographie en phase liquide haute
performance |
|
chromatographie en phase gazeuse. |
|
|
|
|
|
|
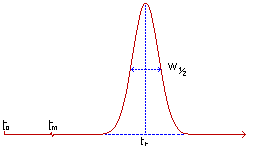
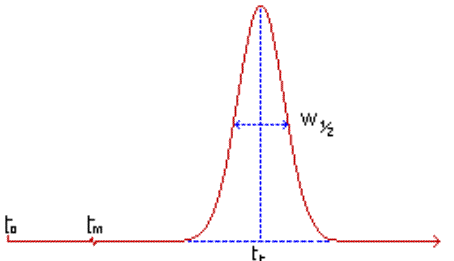
Temps mort tm : temps mis par un
composé non retenu par la phase stationnaire de la colonne, pour parcourir
le trajet entre l'entrée et la sortie de la colonne (temps passé dans la
phase mobile). |
|
Temps de début d’injection : t0 |
|
|
|
|
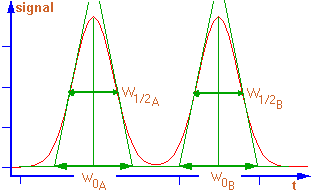
Temps de rétention tr : temps mis par
les molécules d'un composé à analyser (soluté) pour parcourir le trajet
entre l'entrée et la sortie de la colonne. C'est le temps total passé dans
la colonne. |
|
La surface d'un pic est fonction de la quantité
de constituant dont il est la trace. |
|
|
|
|
|
|
|
|
En chromatographie, chaque composé présentant
une certaine affinité pour la phase stationnaire Cs, passera
tour à tour de la phase mobile (Cm)à la phase stationnaire et
inversement. |
|
Leur affinité est traduite par un coefficient
dit de partage |
|
|
|
|
on définit un plateau théorique comme étant une
portion de colonne d'où les 2 phases sortent en équilibre thermodynamique
entre elles. |
|
Le nombre de plateaux théoriques Nth représente
l'efficacité de la colonne pour chaque composé. |
|
|
|
|
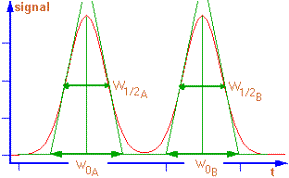
2 constituants sortent à des temps proches, leurs
pics risquent de se chevaucher. En optimisant les conditions analytiques,
il est possible d'améliorer l'allure du chromatogramme. |
|
Le paramètre de résolution R quantifie la
qualité de cette séparation. |
|
|
|
|
|

 Commentaires
Commentaires{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
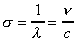{kind=link}
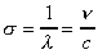{kind=link}
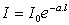{kind=link}
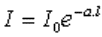{kind=link}
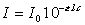{kind=link}
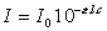{kind=link}
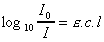{kind=link}
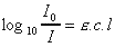{kind=link}
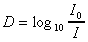{kind=link}
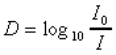{kind=link}
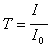{kind=link}
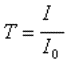{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
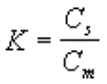{kind=link}
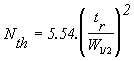{kind=link}
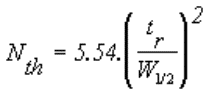{kind=link}
{kind=link}
{kind=link}
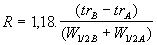{kind=link}
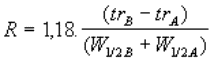{kind=link}